

Read original article at Business Wire
Circle Pharma, in collaboration with the laboratory of Violeta Serra, Ph.D., at Vall d’Hebron Institute of Oncology (VHIO) in Barcelona, Spain, recently unveiled preclinical data demonstrating the efficacy of Circle’s oral Cyclin A/B inhibitor macrocycles in patient-derived breast cancer tumor models. This significant development was presented at the 2023 AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics
The study in 40 breast cancer cell lines identified an association between hallmark pathway scores in E2F, G2/M, and mitotic spindle pathways and sensitivity to Cyclin A/B inhibition. Most notably, multiple triple-negative breast cancer (TNBC) cell lines and luminal B breast cancer cell lines displayed sensitivity to Cyclin A/B inhibition. These findings were instrumental in selecting TNBC and luminal breast cancer patient-derived tumor models for subsequent in vivo efficacy studies. In these in vivo studies, oral administration of Circle’s Cyclin A/B inhibitor led to substantial tumor regression in both TNBC and luminal breast cancer models, with complete response observed in a luminal B breast cancer model derived from a patient whose tumor had previously progressed following multiple treatments, including with a CDK4/6 inhibitor.
Breast cancer is the second leading cause of cancer-related deaths in women globally. Triple negative breast cancer, which accounts for 10-20% of all breast cancer cases, is an aggressive and challenging subtype of breast cancer, associated with poor prognosis and high risk of relapse1. Because TNBC lacks specific receptors (estrogen, progesterone, or HER2) that some other breast cancers have, many newer therapies used to target these receptors in other forms of breast cancer are not effective. Luminal breast cancers are hormone receptor-positive and generally less aggressive than TNBC. Luminal breast cancers can be further divided into two subtypes: luminal A (HR+/ER+/HER2+) and luminal B (HR+/PR+/HER2-). These two subtypes make up about 70% of all breast cancer cases2.
1 American Society of Clinical Oncology. (2017 May) Triple-Negative Breast Cancer: Current Practice and Future Directions. Ricardo L.B. Costa and William J. Gradishar.
2 Nature. (2000; 406) Molecular portraits of human breast tumours. Perou CM, Sorlie T, Eisen MB, et al.
Read original article at Business Wire
SAN FRANCISCO, Calif.–(BUSINESS WIRE)–Circle Pharma announced today that two poster presentations at the American Association for Cancer Research (AACR) Annual Meeting held April 14-19, in Orlando, Florida, highlight promising pre-clinical data of its first-in-class orally bioavailable macrocyclic cyclin A/B inhibitors targeting intractable cancers. Circle expects to advance its cyclin A/B inhibitor program into IND-enabling studies later this year and subsequently into clinical development for testing in a range of cancer types, including SCLC.
The data presented by Circle Pharma shows activity across a wide range of human tumor cell lines and tumor regression in xenograft models of small cell lung carcinoma (SCLC) and ovarian cancer. Pre-clinical development of orally bioavailable macrocycles with dual cyclin A and B inhibitory activity drive synthetic lethality in multiple tumor types. The compounds are shown to be well-tolerated in mice with no observed body weight loss, neutropenia, or depletion of bone marrow across all dose regimens. In vitro studies showed that target engagement in cells leads to displacement of E2F1 from Cyclin A:CDK2 and Myt1 from Cyclin B:CDK1, and the compounds induce DNA damage, G2/M arrest, and apoptosis. The presentation further showed that sensitivity in SCLC cell lines is correlated with RB dysfunction and E2F1 target gene expression.
In addition, studies conducted by the laboratory of Matthew Oser at Dana Farber Cancer Institute (DFCI) validate that cyclin A/B inhibitors induce mitotic arrest and apoptosis in SCLC cell lines. The researchers deployed a genome wide CRISPR/Cas9 positive selection screen to identify that activation of the mitotic spindle assembly checkpoint (SAC) complex by the kinase MSP1 is a dominant mechanism for selective cancer cell killing by the cyclin A/B inhibitors. Dr. Oser’s laboratory at DFCI was sponsored by Circle Pharma.
| Program | Presentation Details |
|---|---|
| Cyclin A/B | Abstract Number: 1559 Abstract Title: Mechanisms responsible for hypersensitivity of small cell lung cancers to novel cyclin A/B RxL macrocyclic peptide inhibitors Session Title: Experimental and Molecular Therapeutics Session Type: Poster |
| Cyclin A/B | Abstract Number: 1560 Abstract Title: Orally bioavailable macrocycles that target cyclins A and B RxL motifs cause tumor regression in xenograft models and in vitro show activity across multiple cancer types Session Title: Experimental and Molecular Therapeutics Session Type: Poster |
About Circle Pharma, Inc.
Circle is a biopharmaceutical company advancing the discovery and development of intrinsically cell-permeable macrocycles that can be delivered by multiple routes, including oral administration. Circle’s MXMO™ platform combines structure-based rational drug design and advanced synthetic chemistry to develop first-in-class macrocycle therapeutics for challenging targets to address unmet clinical needs. Circle’s macrocycle can address both intra- and extra-cellular therapeutic targets and is applicable across a wide range of serious diseases; the company is initially focusing its development efforts on intracellular protein-protein interactions that are key drivers in cancer but have remained elusive to other treatment modalities. Circle is headquartered in South San Francisco and has raised $160 million to date from leading life sciences investors including The Column Group, Nextech, Pfizer and Eli Lilly.
To learn more about Circle Pharma please visit www.circlepharma.com.
Contacts
Circle Pharma Media Contact:
Eleanor Lim
650.825.4099
info@circlepharma.com
Read original article at Business Wire
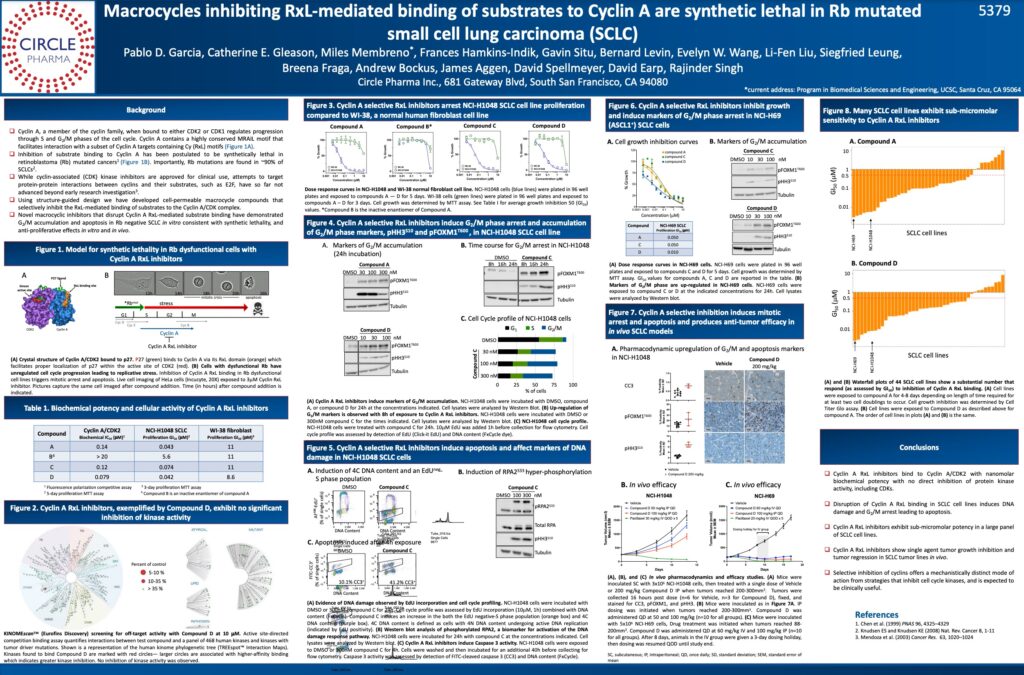
The Company’s presentation provided details of its progress towards structure-guided macrocycles that inhibit the protein-protein interaction between the cyclin A:CDK2 complex and key substrates that are phosphorylated by this complex. Inhibition of Cyclin A substrate binding has been postulated to be synthetic lethal in Rb mutated cancers. The data presented included evidence that macrocycle inhibitors of cyclin A induce G2/M arrest and apoptosis in small cell lung cancer (SCLC) cell lines and have anti-tumor efficacy in SCLC xenograft animal models. Circle plans to advance its cyclin A inhibitor program to the clinic for testing in a range of cancer types, including SCLC where Rb mutations are highly prevalent.
The presentation was made as part of the Mechanisms of Drug Action / Experimental and Molecular Therapeutics session at the AACR meeting, Abstract No. 5379.
Introduction
Cyclic peptide natural products and their synthetic mimics have gained prominence as potential sources of next‐generation therapeutics and biological probes [1–4]. The size and structural complexity of these compounds sets them apart from common synthetic drugs, allowing them to access the “undruggable” target space beyond enzymatic active sites and receptor binding pockets to include activities against a variety of nontraditional targets [4]. Although cyclic peptides have molecular weights and polar group counts that exceed the typical parameters for “drug‐likeness” [5, 6], many are capable of penetrating cells by passive diffusion, and some, such as cyclosporin A, are orally bioavailable [7]. Passive diffusion offers an advantage over other forms of permeation such as paracellular transport, carrier‐mediated transport, and active non‐receptor‐mediated uptake (e.g., micropinocytosis) because the ability to cross the membrane is dictated by the intrinsic properties of the molecule (e.g., molecular weight, number of intramolecular hydrogen bonds (IMHBs), polar surface area, flexibility, lipophilicity) rather than those of the target tissue or cellular physiology (e.g., size of tight junctions, type of transport proteins, invagination of the membrane in response to surface assemblies). Thus, these natural products may provide insights into the requirements for optimizing the ADME properties of large macrocycles.
Further, the stereochemical and conformational complexity of cyclic peptides serves as a model for the design of synthetic scaffolds capable of modulating challenging biological targets such as protein–protein interactions and allosteric binding sites in both extracellular and intracellular space [8–13]. Despite this potential, few efforts have been made to systematically assess the relationship between the structure, pharmacokinetics, and bioactivity of cyclic peptide natural products. This is due in part to the limited number of known passively permeable cyclic peptide and cyclic peptide/polyketide natural products. Therefore the generality of cyclic peptides as orally bioavailable bioactive scaffolds remains an open question.
The few studies that have systematically explored the relationships between structure and permeability in cyclic peptides have been limited to a small subset of methylated (1) [14] and non‐methylated (2) [15] cyclic hexapeptide scaffolds that bear resemblance to baceridin (3) [16], segelin I (4) [17], and the nocardiamide (5/6) [18] cyclic hexapeptide natural products (Figure 5.1 and Chapter 3) [19–22].
The work of Lokey and Jacobson [23–27], Fairlie and Craik [28–30], and others has begun to elucidate the structure– permeability relationships of more complex natural products, but the vast majority of these studies have been limited to cyclic penta‐ and hexapeptides with no observed bioactivity. Thus, the new frontier in understanding structure–permeability relationships in cyclic peptides has moved to the chemical space that encompasses macrocycles of higher molecular weight [31, 32], greater structural complexity, and significant bioactivity.
Here, we first discuss the two‐dimensional and three dimensional (3D) structures of known passively permeable cyclic peptide natural products and then highlight recently discovered cyclic peptide natural products with notable bioactivity that could serve as starting structures for future systematic structural studies to optimize oral absorption.
October 2017
Wiley Publishers
Read online book
by Markus Boehm, Kevin Beaumont, Rhys M Jones, Amit S. Kalgutkar, Liying Zhang, Karen Atkinson, Guoyun Bai, Janice A. Brown, Heather Eng, Gilles H. Goetz, Brian R Holder, Bhagyashree Khunte, Sarah Lazzaro, Chris Limberakis, Sangwoo Ryu, Michael J Shapiro, Laurie Tylaska, Jiangli Yan, Rushia Turner, Siegfried S. F. Leung, Mahesh Ramaseshan, David A. Price, Spiros Liras, Matthew P Jacobson, David J. Earp, R. Scott Lokey, Alan M Mathiowetz, and Elnaz Menhaji-Klotz
Discovery of potent and orally bioavailable macrocyclic peptide-peptoid hybrid CXCR7 modulators.
ABSTRACT: The chemokine receptor CXCR7 is an attractive target for a variety of diseases. While several small molecule modulators of CXCR7 have been reported, peptidic macrocycles may provide advantages in terms of potency, selectivity, and reduced off-target activity. We produced a series of peptidic macrocycles that incorporate an N-linked peptoid functionality where the peptoid group enabled us to explore side chain diversity well beyond that of natural amino acids. At the same time, computational calculations and experimental assays were used to track and reduce polarity while closely monitoring physicochemical properties. This strategy led to the discovery of macrocyclic peptide-peptoid hybrids with high CXCR7 binding affinities (Ki < 100 nM) and measurable passive permeability (Papp > 5 x 10-6 cm/sec). Moreover, bioactive peptide 25 (Ki = 9 nM) achieved oral bioavailability of 18% in rats, which was commensurate with the observed plasma clearance values upon intravenous administration.
October 18, 2017
Journal of Medicinal Chemistry
Read online article
by Matthew R. Naylor, Andrew T. Bockus, Maria-Jesus Blanco, Scott Lokey
Highlights
As interest in protein–protein interactions and other previously-undruggable targets increases, medicinal chemists are returning to natural products for design inspiration toward molecules that transcend the paradigm of small molecule drugs. These compounds, especially peptides, often have poor ADME properties and thus require a more nuanced understanding of structure-property relationships to achieve desirable oral bioavailability. Although there have been few clinical successes in this chemical space to date, recent work has identified opportunities to introduce favorable physicochemical properties to peptidic macrocycles that maintain activity and oral bioavailability.
May 29, 2017
Elsevier
Read online article
by Cameron R. Pye, William M. Hewitt, Joshua Schwochert, Terra D. Haddad, Chad E. Townsend, Lyns Etienne, Yongtong Lao, Chris Limberakis, Akihiro Furukawa, Alan M. Mathiowetz, David A. Price, Spiros Liras, and R. Scott Lokey
Macrocyclic peptides are considered large enough to inhibit “undruggable” targets, but the design of passively cell-permeable molecules in this space remains a challenge due to the poorly understood role of molecular size on passive membrane permeability. Using split-pool combinatorial synthesis, we constructed a library of cyclic, per-N-methlyated peptides spanning a wide range of calculated lipohilicities (0 < AlogP < 8) and molecular weights (∼800 Da < MW < ∼1200 Da). Analysis by the parallel artificial membrane permeability assay revealed a steep drop-off in apparent passive permeability with increasing size in stark disagreement with current permeation models. This observation, corroborated by a set of natural products, helps define criteria for achieving permeability in larger molecular size regimes and suggests an operational cutoff, beyond which passive permeability is constrained by a sharply increasing penalty on membrane permeation…
January 6, 2017
Journal of Medicinal Chemistry
Read online article
by Akihiro Furukawa, Chad E. Townsend, Joshua Schwochert, Cameron R. Pye, Maria A. Bednarek, and R. Scott Lokey
Synthetic and natural cyclic peptides provide a testing ground for studying membrane permeability in nontraditional drug scaffolds. Cyclic peptomers, which incorporate peptide and N-alkylglycine (peptoid) residues, combine the stereochemical and geometric complexity of peptides with the functional group diversity accessible to peptoids. We synthesized cyclic peptomer libraries by split-pool techniques, separately permuting side chain and backbone geometry, and analyzed their membrane permeabilities using the parallel artificial membrane permeability assay. Nearly half of the side chain permutations had permeability coefficients (Papp) > 1 × 10–6 cm/s. Some backbone geometries enhanced permeability due to their ability to form more stable intramolecular hydrogen bond networks compared with other scaffolds. These observations suggest that hexameric cyclic peptomers can have good passive permeability even in the context of extensive side chain and backbone variation, and that high permeability can generally be achieved within a relatively wide lipophilicity range.
October 3, 2016
Journal of Medicinal Chemistry
Read online article
by Joshua Schwochert, Yongtong Lao, Cameron R. Pye, Matthew R. Naylor, Prashant V. Desai, Isabel C. Gonzalez Valcarcel, Jaclyn A. Barrett, Geri Sawada, Maria-Jesus Blanco, and R. Scott Lokey
Cyclic peptide (CP) natural products provide useful model systems for mapping “beyond-Rule-of-5” (bRo5) space. We identified the phepropeptins as natural product CPs with potential cell permeability. Synthesis of the phepropeptins and epimeric analogues revealed much more rapid cellular permeability for the natural stereochemical pattern. Despite being more cell permeable, the natural compounds exhibited similar aqueous solubility as the corresponding epimers, a phenomenon explained by solvent-dependent conformational flexibility among the natural compounds. When analyzing the polarity of the solution structures we found that neither the number of hydrogen bonds nor the total polar surface area accurately represents the solvation energies of the high and low dielectric conformations. This work adds to a growing number of natural CPs whose solvent-dependent conformational behavior allows for a balance between aqueous solubility and cell permeability, highlighting structural flexibility as an important consideration in the design of molecules in bRo5 chemical space.
June 6, 2016
ACS Medicinal Chemistry Letter
Read online article
